



 DONATE NOW
DONATE NOWThis weekend’s Saturday Night Live skit about the recent historic approvals of potentially curative gene therapies for sickle cell disease is distasteful at best and harmful at worst. Earlier this month, the Food and Drug Administration approved groundbreaking new treatments that could change the lives of thousands. SNL chose to cast a spotlight on this news with a tone-deaf skit depicting a workplace Yankee Swap event in which one of the gifts is the “cure” for sickle cell disease. It is given to an African American character, who quickly trades it for a “Boogie Woogie Santa Claus” toy. The rest of the skit consists of the white characters trying to convince their two Black co-workers to choose the cure over the other Yankee Swap gifts. Their attempts are unsuccessful.
We are disappointed that Saturday Night Live chose to trivialize this landmark moment in history during their program. More than 100,000 people in the United States and millions globally are impacted by this devastating disease, and yet it is one of the few debilitating conditions that you will find people joking about on television. Earlier this year, sickle cell disease was the subject of a lame and insensitive attempt at humor on the HBO Max show Velma and, shortly thereafter, as a quasi-joke-insult by comedian D.L. Hughley on The Daily Show. Some may argue that these references are “just jokes,” but for those impacted by this disease, it is no laughing matter.
Jokes like these undermine the seriousness of this condition. Sickle cell disease (SCD) is an inherited blood disorder and rare disease that affects red blood cells. When these red blood cells become sickle-shaped, or crescent-shaped, they block blood flow to the affected part of the body, causing irreversible organ and tissue damage. When this happens, individuals with sickle cell can suffer from intractable, crippling acute pain called a “crisis” and are at elevated risk for strokes, damage to affected tissue, and all too often, an early death. In fact, a recent study showed the median age of death of those suffering from chronic sickle disease complications was only 43 years.
SNL’s treatment of race in the Yankee Swap skit also misses the mark. Part of the “humor” revolves around the common myth that only Black people can have sickle cell disease. While it does disproportionately impact the Black community, sickle cell does not discriminate. People of all ethnic backgrounds can inherit the disease. In the United States, Hispanic and Latino populations have the second highest incidence, but Asian, Indian, Native American and – yes – White people, can all be born with the disease. On a global scale, sickle cell disease affects people from countries around the world, including Italy, India, the United Kingdom and Jamaica. One doesn’t develop sickle cell disease, nor can one “catch it.” Individuals are born with it, and there is no universal cure.
Why are we joking about a disease as serious as this one? Many people don’t understand the devastating reality of the condition. The onset of sickle cell pain is sudden and debilitating. A pain crisis is relentless and can last for hours or for days. It has been described as feeling like you are walking on hot coals or like shards of glass are traveling through your veins. Far too often, when individuals living with sickle cell disease, or “warriors” as they call themselves, are in crisis and seek medical care in some emergency departments, they face long waiting periods, are accused of exaggerating symptoms for attention, and far, far too often are characterized and treated as if they are drug seekers.
For physicians who are knowledgeable about sickle cell disease and experienced in caring for those living with it, their ability to prescribe the very drugs that will help their patients is hampered by current federal regulations put in place to address the opioid crisis thus limiting how these drugs can be used in cases such as sickle cell. Layer on issues of health care inequity, discrimination and limited access to consistent, comprehensive quality care and the word “crisis” takes on new meaning.
Community-based organizations, such as the 50-plus members of the Sickle Cell Disease Association of America, Inc., spanning 29 states, are on the ground and focused on providing support, resources, and services to serve more than 500,000 children and adults living with or impacted by sickle cell disease.
Sickle cell disease also puts a strain on caregivers and family members, who must fit trips to the emergency room, doctors’ appointments and sick days into the rigors of daily life. Parents of children with sickle cell may lose wages, promotion opportunities or jobs as they try to support their family while attending to pain crises and their child’s care. This pressure can cause personal and professional instability, compromise mental health and wellness, and, in too many cases we have seen, lead to homelessness.
It is for all of the above, and more, that the Sickle Cell Disease Association of America, Inc., condemns the use of sickle cell disease as a punchline. It demeans and ridicules a condition that people are born with and from which they will face devastating health challenges throughout their lifetimes.
Stereotypes and misinformation reinforced by thoughtless comedy have real-life consequences. Sickle cell patients struggle daily to be taken seriously—in school, at work and even playing sports. As we work to change the perception of sickle cell and increase education surrounding this condition, insensitive and inappropriate jokes like these demean, marginalize and disrespect those living with the disease. They work against progress and contribute to the spread of misinformation. As a society, we must do better and treat rare diseases and the people who live with them with the respect they deserve.
Sickle cell is not a joke.
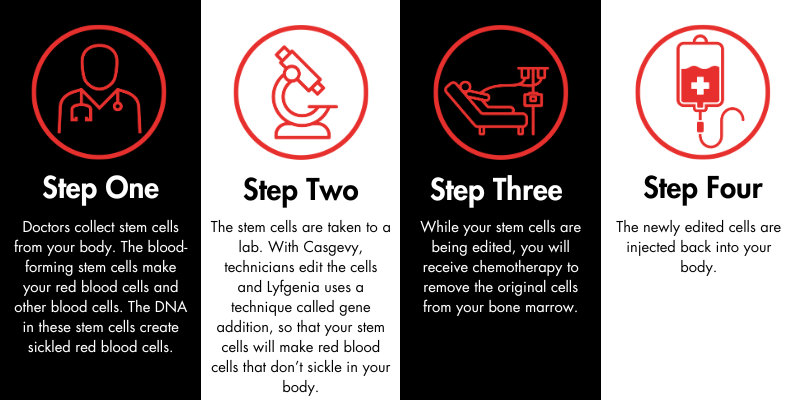

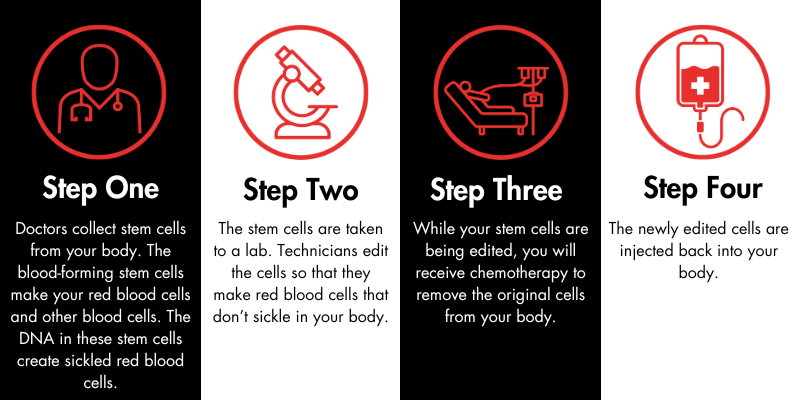
 The Sickle Cell Disease Association of America, Inc., (SCDAA) is saddened to hear the news of the passing of Dr. Lennette Benjamin. Dr. Benjamin was a trailblazing physician who made many outstanding contributions to the sickle cell community. She was one of the first to establish a “day hospital” as an alternative to the emergency room for pain management – an approach that is today recognized as a best practice in care.
The Sickle Cell Disease Association of America, Inc., (SCDAA) is saddened to hear the news of the passing of Dr. Lennette Benjamin. Dr. Benjamin was a trailblazing physician who made many outstanding contributions to the sickle cell community. She was one of the first to establish a “day hospital” as an alternative to the emergency room for pain management – an approach that is today recognized as a best practice in care.